Used softwares
Data source
$ axel ftp://ftp.ncbi.nlm.nih.gov/genomes/refseq/bacteria/assembly_summary.txt
Reformat
$ cat assembly_summary.txt | sed 1d | sed '1s/^# //' \
| sed 's/"/$/g' > assembly_summary.tsv
$ csvtk stats -t assembly_summary.tsv
file num_cols num_rows
assembly_summary.tsv 22 83,308
# aligned view
$ csvtk -t pretty assembly_summary.tsv > assembly_summary.tsv.pretty
Headers
$ csvtk headers -t assembly_summary.tsv
# assembly_summary.tsv
1 assembly_accession
2 bioproject
3 biosample
4 wgs_master
5 refseq_category
6 taxid
7 species_taxid
8 organism_name
9 infraspecific_name
10 isolate
11 version_status
12 assembly_level
13 release_type
14 genome_rep
15 seq_rel_date
16 asm_name
17 submitter
18 gbrs_paired_asm
19 paired_asm_comp
20 ftp_path
21 excluded_from_refseq
22 relation_to_type_material
Strains with Complete Genome
$ cat assembly_summary.tsv \
| csvtk grep -t -f assembly_level -i -p "Complete Genome" \
| wc -l
7742
Number of species
Counting by species_taxid.
$ cat assembly_summary.tsv \
| csvtk cut -t -f species_taxid \
| csvtk freq -t \
| sed 1d | wc -l
12445
Counting by organism_name (not precise)
$ cat assembly_summary.tsv \
| csvtk cut -t -f organism_name \
| cut -d ' ' -f 1,2 \
| csvtk freq -t \
| sed 1d | wc -l
7086
Most sequenced species
$ cat assembly_summary.tsv \
| csvtk cut -t -f organism_name \
| cut -d ' ' -f 1,2 \
| csvtk freq -t -n -r | head -n 20 | csvtk pretty -t
organism_name frequency
Staphylococcus aureus 7441
Streptococcus pneumoniae 7257
Salmonella enterica 6903
Escherichia coli 5385
Mycobacterium tuberculosis 5088
Pseudomonas aeruginosa 2230
Acinetobacter baumannii 1947
Klebsiella pneumoniae 1798
Mycobacterium abscessus 1375
Listeria monocytogenes 1324
Shigella sonnei 958
Streptococcus suis 955
Clostridioides difficile 901
Campylobacter jejuni 899
Streptococcus agalactiae 867
Campylobacter coli 802
Neisseria meningitidis 790
Vibrio parahaemolyticus 685
Helicobacter pylori 659
Well organized directory structures in Bioinformatics project make the analysis more scalable and reproducible, and are easy for parallelization.
Here’s my way.
Directory raw contains all paired-ends reads files, which are carefully
named with same suffixes: _1.fastq.gz for read 1 and _2.fastq.gz for read 2.
$ ls raw | head -n 4
delta_G_9-30_1.fastq.gz
delta_G_9-30_2.fastq.gz
WT_9-30_1.fastq.gz
WT_9-30_2.fastq.gz
...
I use a python script cluster_files to cluster paired-end reads by samples:
$ cluster_files -p '(.+?)_[12]\.fastq\.gz$' raw/ -o raw.cluster
$ tree
.
├── raw
│ ├── delta_G_9-30_1.fastq.gz
│ ├── delta_G_9-30_2.fastq.gz
│ ├── WT_9-30_1.fastq.gz
│ └── WT_9-30_2.fastq.gz
└── raw.cluster
├── delta_G_9-30
│ ├── delta_G_9-30_1.fastq.gz -> ../../raw/delta_G_9-30_1.fastq.gz
│ └── delta_G_9-30_2.fastq.gz -> ../../raw/delta_G_9-30_2.fastq.gz
└── WT_9-30
├── WT_9-30_1.fastq.gz -> ../../raw/WT_9-30_1.fastq.gz
└── WT_9-30_2.fastq.gz -> ../../raw/WT_9-30_2.fastq.gz
A framework for safely handling Ctrl-C in Golang.
cancel := make(chan struct{})
// do some jobs
chWaitJobs := make(chan int)
go func(cancel chan ) { // one example
// do jobs that may take long time
LOOP:
for {
select {
case <-cancel:
break LOOP
default:
}
// do something
}
<- chWaitJobs
}(cancel)
chExitSignalMonitor := make(chan struct{})
cleanupDone := make(chan int)
signalChan := make(chan os.Signal, 1)
signal.Notify(signalChan, os.Interrupt)
go func() {
select {
case <-signalChan:
log.Criticalf("received an interrupt, cleanning up ...")
// broadcast cancel signal, so other jobs can exit safely
close(cancel)
// cleanning work
cleanupDone <- 1
return
case <-chExitSignalMonitor:
cleanupDone <- 1
return
}
}()
// wait jobs done
<-chWaitJobs
close(chExitSignalMonitor)
<-cleanupDone
Reference
- https://nathanleclaire.com/blog/2014/08/24/handling-ctrl-c-interrupt-signal-in-golang-programs/ this method is only for long-time-running service.
See http://bioinf.shenwei.me/seqkit/note/
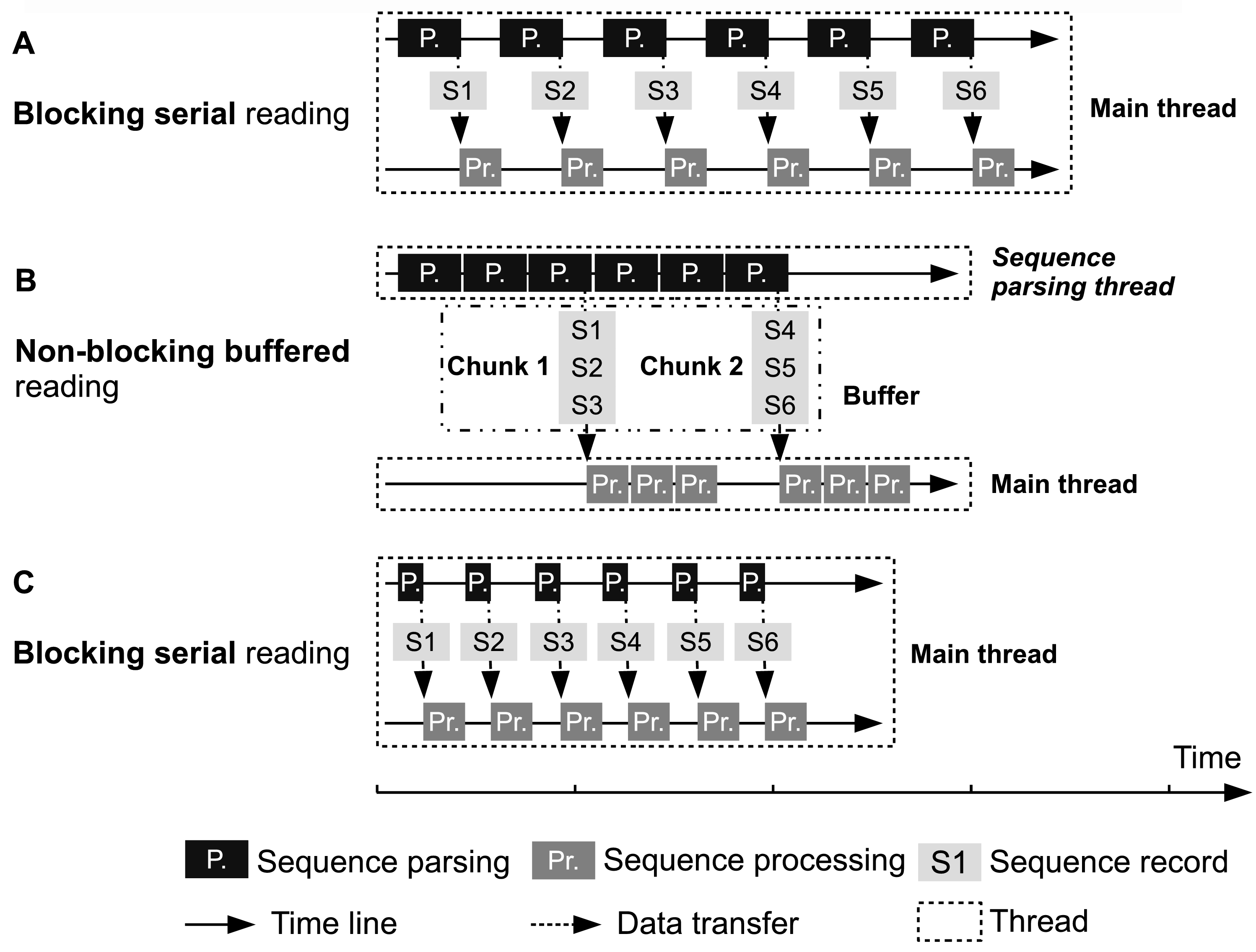
Illustration of FASTA/Q file parsing strategies. (A) and (C) Main thread parses one sequence, waits (blocked) it to be processed and then parses next one. (B) Sequence parsing thread continuously (non-blocked) parses sequences and passes them to main thread. The width of rectangles representing sequence parsing and sequence processing is proportional with running time. Sequence parsing speeds in (A) and (B) are the same, which are both much slower than that in (C). The speeds of sequence processing are identical in (A), (B) and (C). In (B), chunks of sequences in buffer can be processed in parallel, but most of the time the main thread needs to serially manipulate the sequences.
During the past three months, I’ve wrote two tools (SeqKit and csvtk) and extended few packages (bio and util), all in Go language.
Here are some experience I’ve got.
Code organiztion
src # source code
docs # documents
tests # tests
benchmarks # benchmark results
examples # examples
LICENSE
README.md
Version control
- Use git.
- Create a repository on Github.
Documents
- Write annotation for all public variables and functions. Use lint tools to check this.
- Duild a project website (mkdocs/hugo) and host it on github.io.
Tests
Very important!
- Unit Test.
- Cover all functions, especially the frenquently used packages.
- Use test tool of the programming language.
- Function Test.
- Use automated tools, e.g. ssshtest - Stupid Simple (ba)Sh Testing - A functional software testing framwork
Automation
- Packing release files.
- Automated testing.
Language
data types
int(2.3)
const
#define PI 3.1415926
or
const double pi = 3.1415;
I wrote a bioinformatics package in golang,
in which I used a function to check whether a letter(byte) is a valid DNA/RNA/Protein letter.
The easy way is storing the letters of alphabet in a map and check the existance of a letter.
However, when I used go tool pprof to profile the performance,
I found the hash functions (mapaccess2, memhash8, memhash)
of map cost much time (see figure below).
Then I found a faster way: storing letters in a slice,
in detail, saving a letter(byte) at position int(letter) of slice.
To check a letter, just chech the value of slice[int(letter)], non-zero means
valid letter.
[update at 2016-06-02] Two switch versions were also tested.
They were faster than map version, but still slower than slice version.
Besides, it was affected by the number of case sentences in switch,
i.e. the bigger the alphabet size is, the slower it runs.
See the benchmark result:
| Tests | Iterations | Time/operation |
|---|---|---|
| BenchmarkCheckLetterWithMap-4 | 2000000000 | 0.18 ns/op |
| BenchmarkCheckLetterWithSwitch-4 | 1000000000 | 0.02 ns/op |
| BenchmarkCheckLetterWithSwitchWithLargerAlphabetSize-4 | 1000000000 | 0.03 ns/op |
| BenchmarkCheckLetterWithSlice-4 | 2000000000 | 0.01 ns/op |
[updates] I wrote a tool to do the same job and it’s even more powerful.
gTaxon - a fast cross-platform NCBI taxonomy data querying tool, with cmd client and REST API server for both local and remote server. http:/github.com/shenwei356/gtaxon
This post presents a script for fetching taxon information by species name or taxid.
Take home message:
1). using cache to avoid repeatly search
2). object of Entrez.read(Entrez.efetch()) could be treated as list,
but it could not be rightly pickled. Using Json is also not OK.
The right way is cache the xml text.
search = Entrez.efetch(id=taxid, db="taxonomy", retmode="xml")
# data = Entrez.read(search)
## read and parse xml
data_xml = search.read()
data = list(Entrez.parse(StringIO(data_xml)))
3). pickle file was fragile. A flag file could be used to detect whether data is rightly dumped.
4). using multi-threads to accelerate fetching.
Basic
- NumPy - NumPy is the fundamental package for scientific computing with Python
- pandas - pandas is an open source, BSD-licensed library providing high-performance, easy-to-use data structures and data analysis tools for the Python programming language.
- matplotlib - matplotlib is a python 2D plotting library which produces publication quality figures in a variety of hardcopy formats and interactive environments across platforms
- seaborn - Statistical data visualization using matplotlib
Further
- Statsmodels - Statsmodels is a Python module that allows users to explore data, estimate statistical models, and perform statistical tests.
- scikit-learn - Machine Learning in Python
- theano - Theano is a Python library that allows you to define, optimize, and evaluate mathematical expressions involving multi-dimensional arrays efficiently